Topological Crystal Structure Prediction
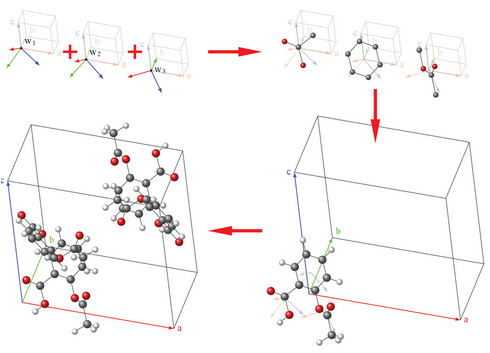
Organic molecular crystals constitute a class of materials of critical importance in numerous industries. Despite the ubiquity of these systems, ability to predict molecular crystal structures starting only from a two-dimensional diagram of the constituent compound(s) remains a significant challenge. Most structure-prediction protocols require a customized interatomic interaction model on which the quality of the results can depend sensitively.
To overcome this problem, a new topological approach was introduced to molecular crystal structure prediction. The approach posits that in a stable structure,molecules are oriented such that principal axes and normal ring plane vectors are aligned with specific crystallographic directions and that heavy atoms occupy positions that correspond to minima of a set of geometric order parameters.
By minimizing an objective function that encodes these orientations and atomic positions, and filtering based on the vdW free volume and intermolecular close contact distributions derived from the Cambridge Structural Database, stable structures and polymorphs for a given crystal can be predicted entirely mathematically without reliance on an interaction model.
