Efficient Molecular Crystal Structure Prediction and Stability Assessment with AIMNet2 Neural Network Potentials
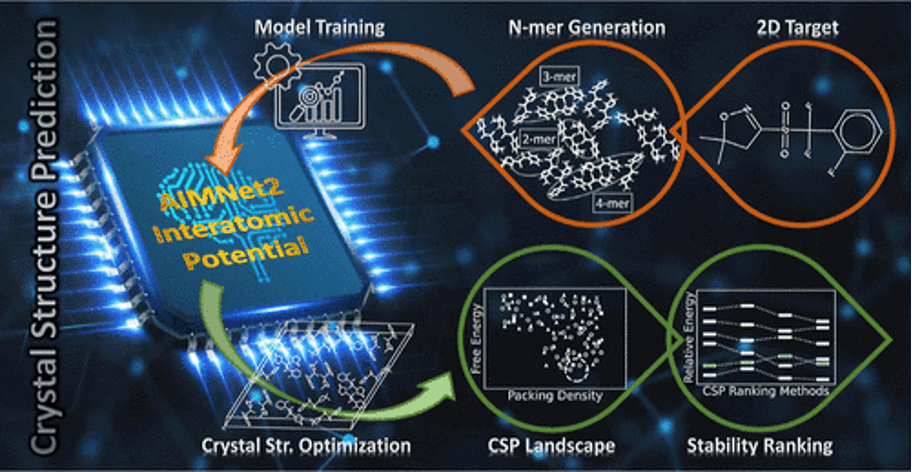
Identifying thermodynamically stable crystal structures remains a key challenge in materials chemistry. Computational crystal structure prediction (CSP) workflows typically rank candidate structures by lattice energy to assess relative stability. Approaches using self-consistent first-principles calculations become prohibitively expensive, especially when millions of energy evaluations are required for complex molecular systems with many atoms per unit cell. Here, an approach that significantly accelerates CSP by training target-specific machine-learned interatomic potentials (MLIPs) is presented. AIMNet2 MLIPs are trained on density functional theory (DFT) calculations of molecular clusters, herein referred to as n-mers. Potentials trained on gas phase dispersion-corrected DFT reference data of n-mers are demonstrated to successfully extend to crystalline environments, accurately characterizing the CSP landscape and correctly ranking structures by relative stability. This methodology effectively captures the underlying physics of thermodynamic crystal stability using only molecular cluster data, avoiding the need for expensive periodic calculations. The performance of target-specific AIMNet2 interatomic potentials is illustrated across diverse chemical systems relevant to pharmaceutical, optoelectronic, and agrochemical applications, demonstrating their promise as efficient alternatives to full DFT calculations for routine CSP tasks.
