Reorganization Energy Predictions with Graph Neural Networks
These results demonstrate the feasibility of reorganization energy predictions on the benchmark QM9 data set without needing DFT-optimized geometries and demonstrate the types of features needed for robust models that work on diverse chemical spaces.
Daniel Tabor
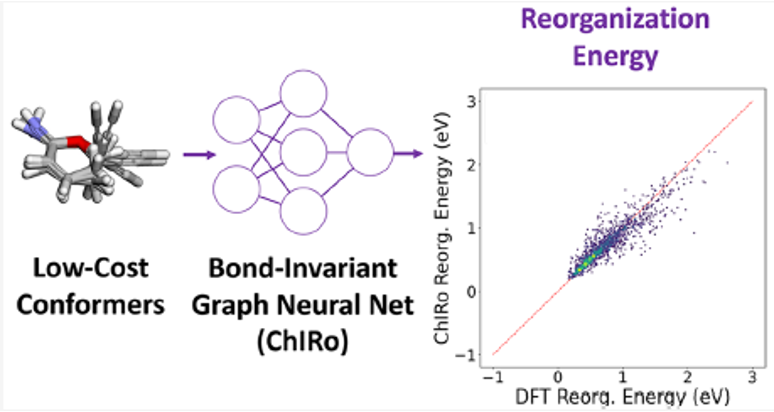
A critical bottleneck for the design of high conductivity organic materials is finding molecules with low reorganization energy. To enable high-throughput virtual screening campaigns for many types of organic electronic materials, a fast reorganization energy prediction method compared to density functional theory is needed. However, the development of low-cost machine-learning-based models for calculating the reorganization energy has proven to be challenging. Here a 3D graph-based neural network (GNN) recently benchmarked for drug design applications, ChIRo, was combined with low-cost conformational features for reorganization energy predictions. By comparing the performance of ChIRo to another 3D GNN, SchNet, evidence was found that the bond-invariant property of ChIRo enables the model to learn from low-cost conformational features more efficiently. Through an ablation study with a 2D GNN, it was found that using low-cost conformational features on top of 2D features informs the model for making more accurate predictions. These results demonstrate the feasibility of reorganization energy predictions on the benchmark QM9 data set without needing DFT-optimized geometries and demonstrate the types of features needed for robust models that work on diverse chemical spaces. Furthermore, it was shown that ChIRo informed with low-cost conformational features achieves comparable performance with the previously reported structure-based model on π-conjugated hydrocarbon molecules. It is expected that this class of methods can be applied to the high throughput screening of high-conductivity organic electronics candidates.